Research

At CICEKLAB, we design and develop machine learning algorithms for the bioinformatics pipeline to improve our understanding of genetic diseases and disorders. Below are some of the recent projects we work on:
RNA Design
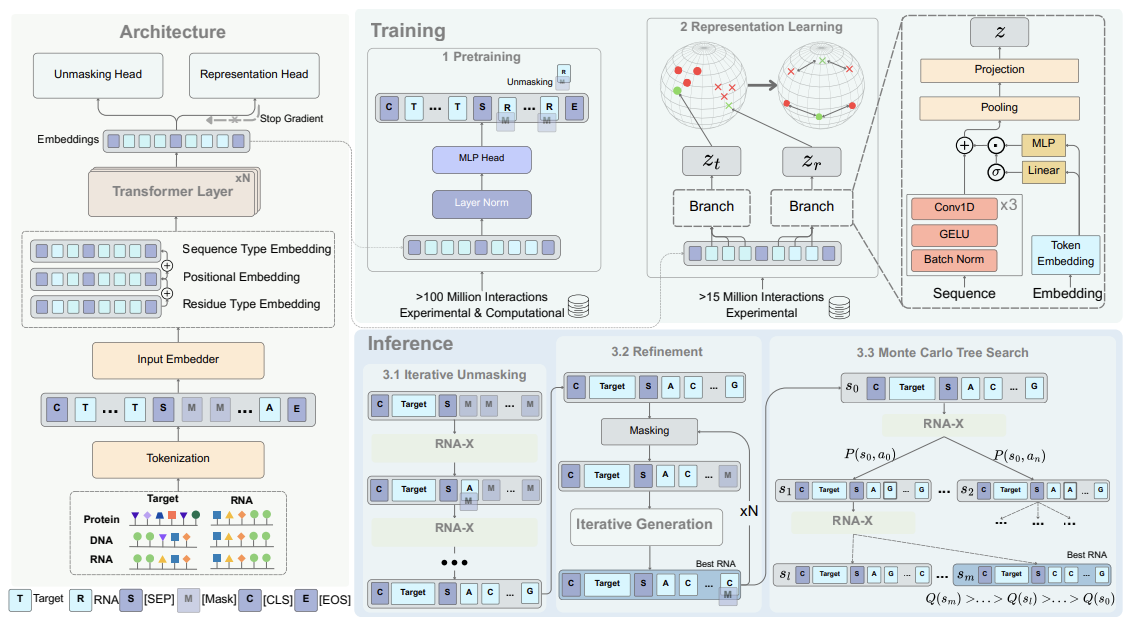
The system model for RNA-X
Our research lab is focused on developing the next generation of RNA foundation models to transform the landscape of therapeutic design. We address the fundamental challenge of designing RNA sequences that can selectively and strongly bind to specific target molecules—a process traditionally hindered by the vastness of the sequence space and the limitations of trial-and-error experimental methods. To solve this, we developed UTRGAN, RNAGEN, RNAtranslator, and recently RNA-X. The latest work, RNA-X is a novel interaction-aware foundation model based on masked language modeling and representation learning. Unlike previous methods restricted primarily to protein targets, our work enables the conditional design of RNA binders targeting proteins, other RNA, and DNA molecules.
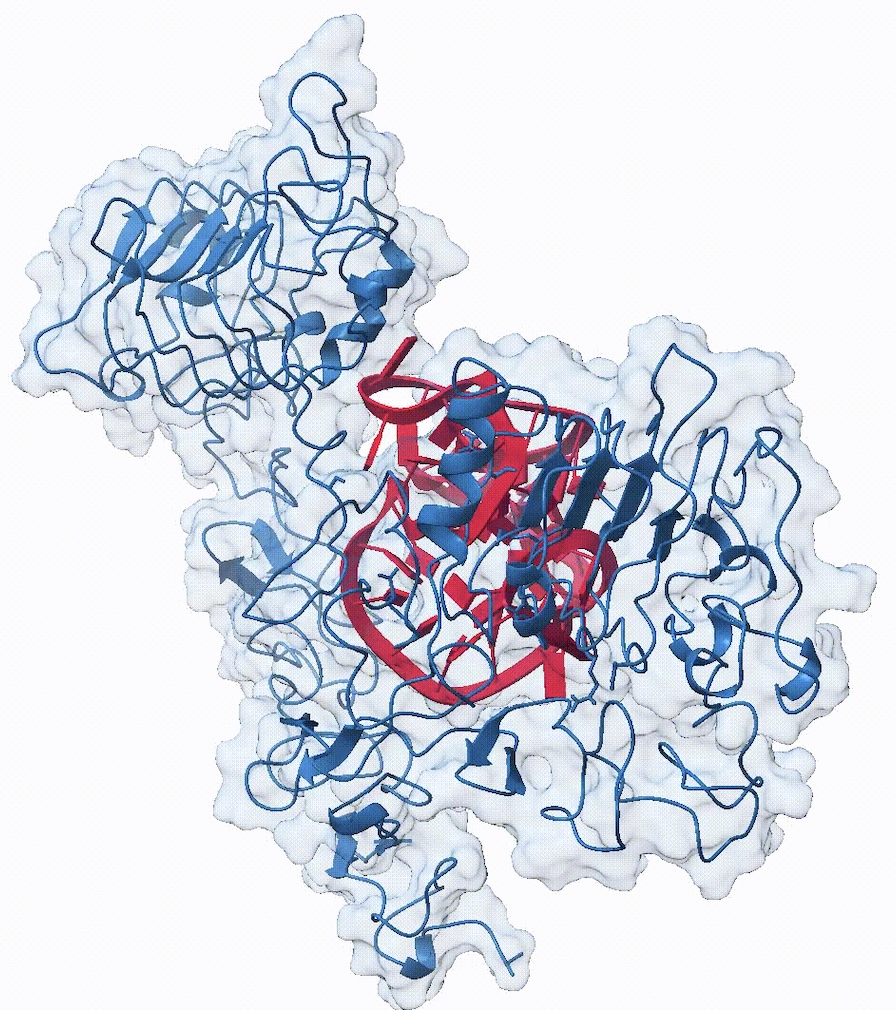
An RNA molecule (red) designed by RNA-X to bind EGFR protein (blue)
A major innovation of our lab is the ability to perform simultaneous multi-target design. As a proof of concept, we successfully designed a novel guide RNA from scratch that binds concurrently to a bacterial DNA sequence and the Cas9 protein, achieving predicted binding energies comparable to wild-type structures. Our methodology integrates generative modeling with a Monte Carlo Tree Search (MCTS) exploration strategy, allowing us to optimize designed sequences for high binding affinity, thermodynamic stability, and structural quality. [cite_start]Despite having significantly fewer parameters than existing models, our optimized embeddings consistently outperform state-of-the-art methods in diverse downstream tasks, such as predicting the efficacy of siRNA and sgRNA.
Copy Number Variation Detection in Challenging Data and Genomic Regions
In our research, we address the longstanding clinical challenge of accurately detecting Copy Number Variants (CNVs) from Whole Exome Sequencing (WES) data. While CNVs are a major cause of genetic disorders, identifying them in WES data has historically been difficult due to low precision and recall on expert-curated gold standards. To solve this, we developed multiple technologies like DECoNT, ExactCN and ECOLE. ECOLE is a deep learning-based caller that utilizes a variant of the transformer architecture to call CNVs on a per-exon basis.
ECOLE is uniquely trained through a multi-stage process: it first learns from high-confidence calls made on matched Whole Genome Sequencing (WGS) samples and is then fine-tuned on a small set of human expert-labeled data via transfer learning. Additionally, the model is capable of detecting variations in cancer samples, such as bladder cancer, without requiring a control sample, making it a powerful and efficient tool for clinical diagnostics.
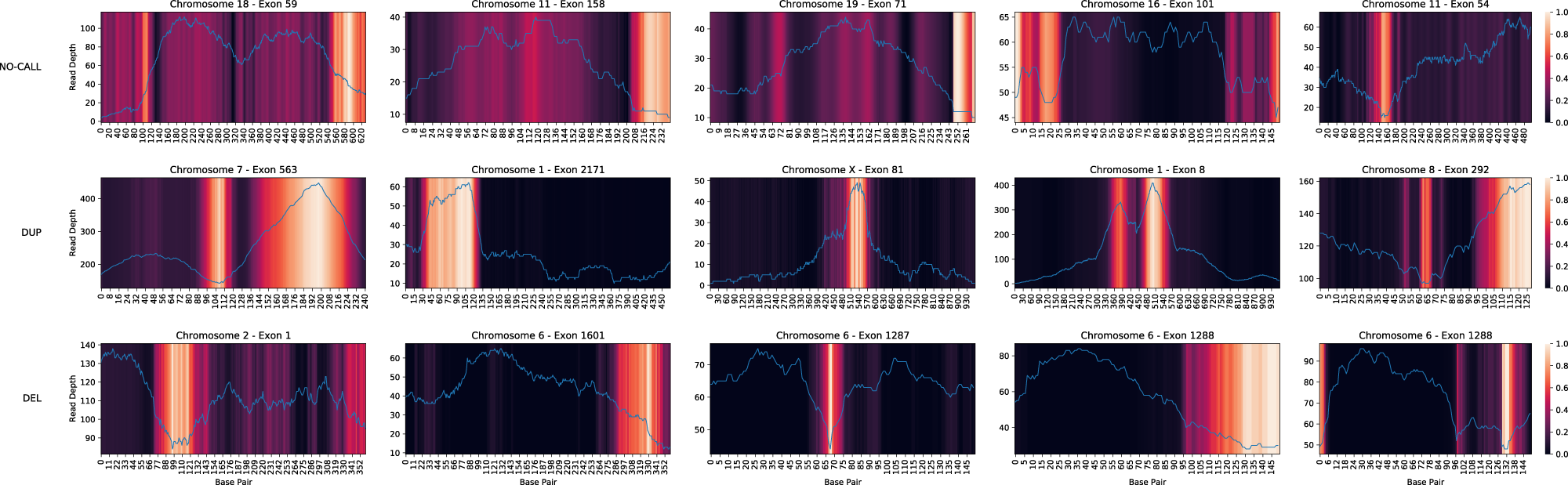
Attention maps for ECOLE show which patterns in the read depth signal result in a CNV call.
We extended this line of work to focus on problems in the WGS data. LYCEUM, which is the first machine learning-based tool specifically designed to detect copy number variants (CNVs) in highly degraded, low-coverage ancient DNA (aDNA). Because ancient genomic samples often suffer from extreme degradation, microbial contamination, and typically low sequencing coverage, conventional CNV algorithms optimized for high-quality modern data often underperform. LYCEUM addresses these challenges using a two-step training strategy: it is first pre-trained on high-coverage whole-genome sequencing data from the 1000 Genomes Project and then fine-tuned on downsampled read depth signals from existing high-coverage ancient samples to adapt to realistic noise levels. Benchmarks demonstrate that LYCEUM maintains state-of-the-art accuracy even at extremely low coverage levels (e.g., 0.05×), and its resulting deletion calls correlate with established demographic histories and patterns of natural selection.
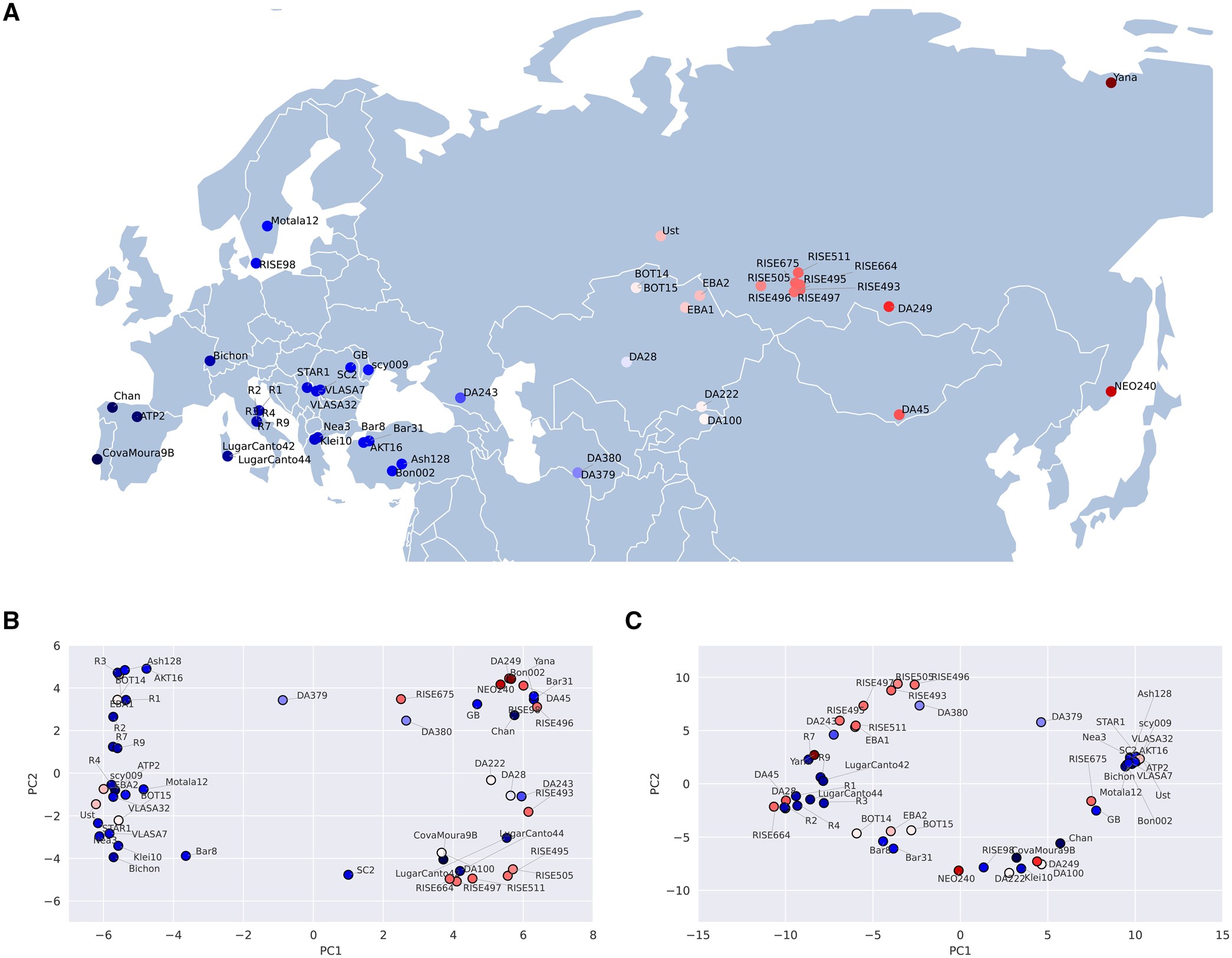
Distribution of segmental deletions. (A) Geographic locations of 50 aDNA samples. (B) and (C) show the visualization of the first two principal components representing genomic distances based on segmental deletion events identified by LYCEUM and CONGA, respectively. Results highly correlate with demographic structure.
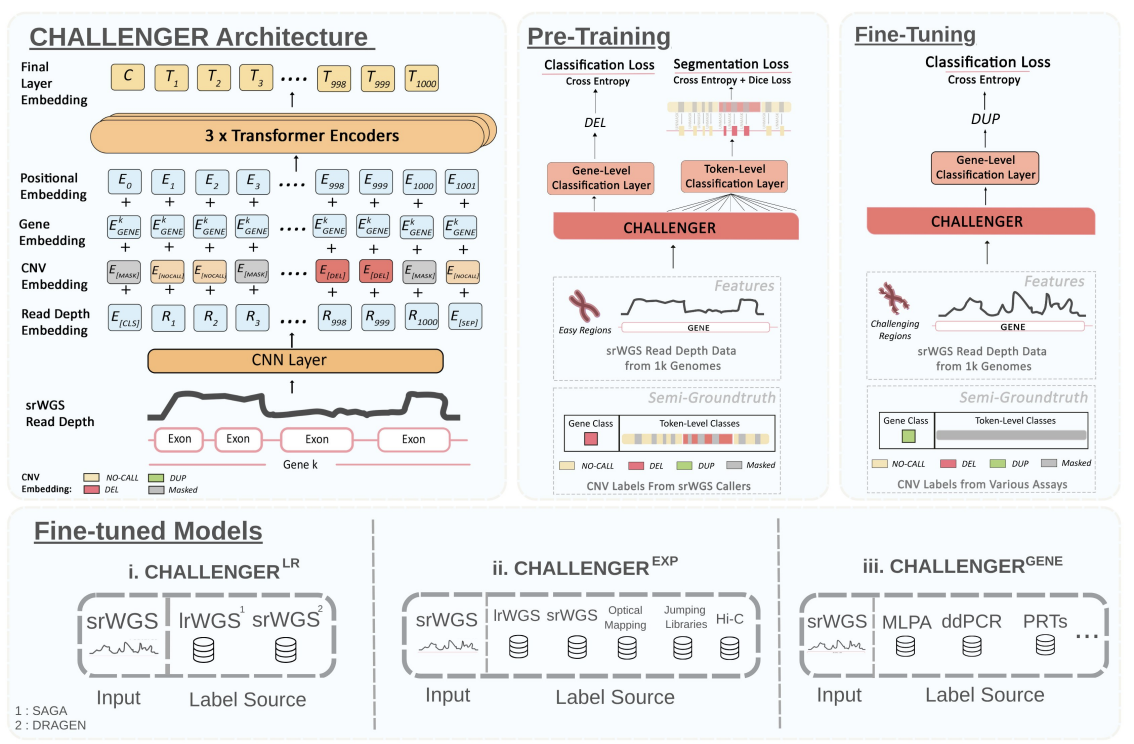
Finally, CNV detection in genomic regions that are difficult to align, such as repetitive, duplicated, or "camouflaged" areas. While long-read sequencing technologies can resolve these structural variants, their high cost often makes them impractical for large-scale clinical use. To solve this, we developed CHALLENGER, a transformer-based deep learning framework that utilizes masked language modeling to detect gene-level CNVs using only standard short-read sequencing data. CHALLENGER is uniquely trained to bridge the gap between sequencing technologies: it is first pre-trained on short-read data and then fine-tuned using high-confidence calls from long-read sequencing, human expert labeling, and experimental validation. This training regime enables the model to pinpoint variants that were previously only accessible via expensive orthogonal technologies or manual expert review. Our results show that CHALLENGER improves the state-of-the-art CNV detection F1-score by 40.8%, successfully capturing over 80% of CNVs in challenging regions that typically require long reads. Additionally, the model is specialized for complex paralog genes, such as SMN1/2 and AMY1/2, allowing for precise, paralog-specific copy number calls.
Ensuring genome data is shared in a privacy preserving way among life scientists
The system model for genom reconstruction attack
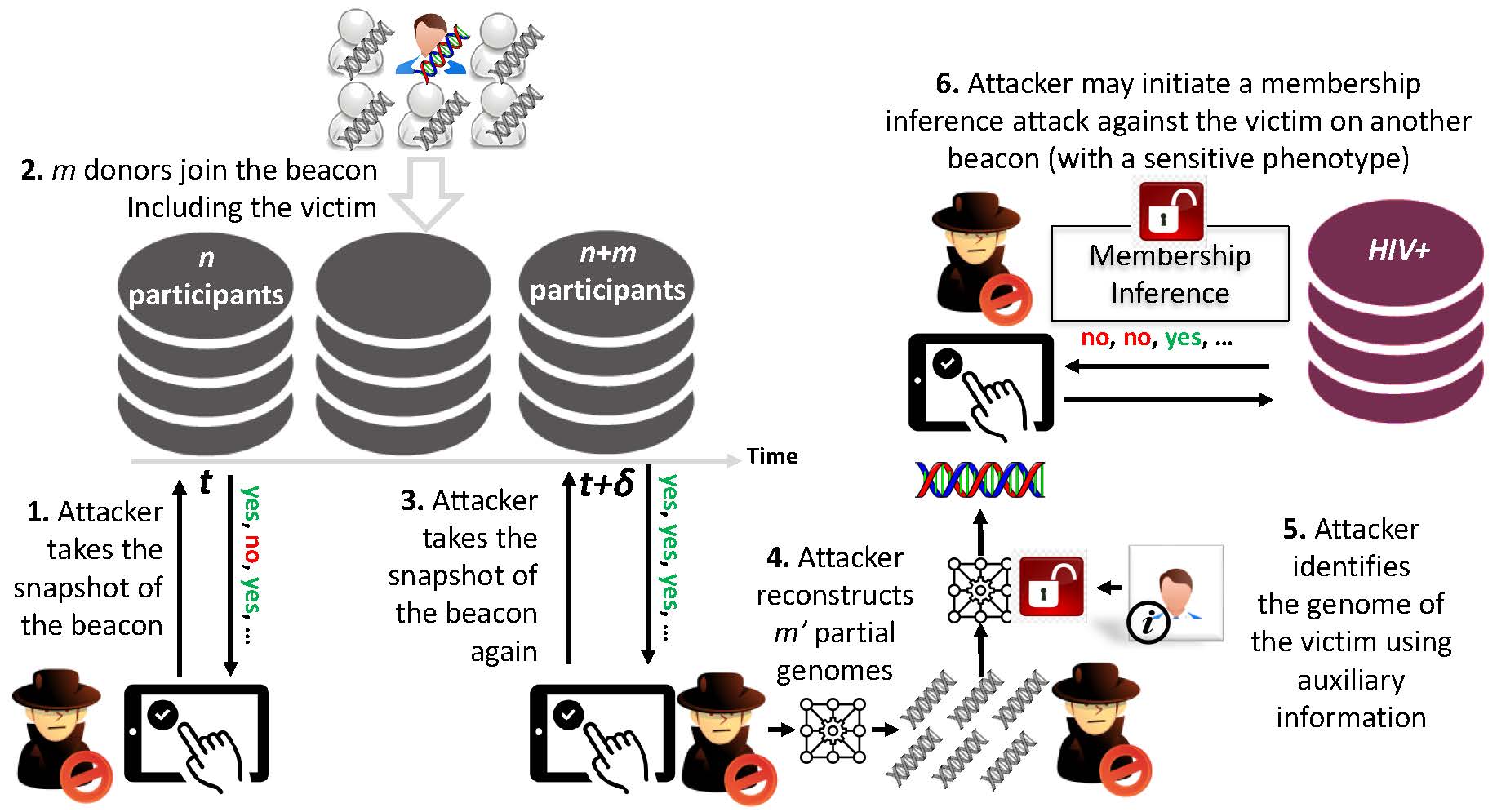
Sharing genome data in a privacy-preserving way stands as a major bottleneck in front of the scientific progress promised by the big data era in genomics. A community-driven protocol named genomic data-sharing beacon protocol has been widely adopted for sharing genomic data. The system aims to provide a secure, easy to implement, and standardized interface for data sharing by only allowing yes/no queries on the presence of specific alleles in the dataset. However, beacon protocol was recently shown to be vulnerable against membership inference attacks.
Our research lab focuses on addressing the critical privacy-utility trade-offs inherent in the sharing of sensitive genomic data. While platforms like the GA4GH Beacon Project enable the global exchange of genetic information to foster medical research, they remain highly vulnerable to sophisticated membership inference and genome reconstruction attacks that can identify individual participants.
We identify such membership inference and genome reconstruction attacks and develop defense mechanisms by using a combination of game theory and reinforcement learning to solve these challenges. Our research involves modeling the interaction between data-sharing services and users as sequential Stackelberg games, allowing the system to anticipate and respond to adversarial behavior in real time. By training RL agents in multi-agent environments, we create adaptive systems capable of distinguishing between legitimate clinical researchers and malicious actors. A core principle of our methodology is scalability and privacy-preserving training; our models are designed to learn from statistical summaries rather than direct access to individual genomes, ensuring that the defense itself does not become a point of data leakage.
Collaborators: Erman Ayday (CWRU), Sinem Sav (Bilkent)
Funding: NIH R01LM013429 (CO-I)